Malm G. von
Döbeln U., Naess K, Ringde´n O. Framsteg om lysosomala sjukdomar
ger hopp om bot och bättringläkartidning
http://www.lakartidningen.se/07engine.php?articleId=11014
ovat
harvinaisia, perinnöllisiä kertymäsairauksia, jotka aiheutuvat
muutoksista lysosomien entsymaattisessa funktiossa. Tällaiset
taudit ovat progredioivia ja johtavat varhaiseen kuolemaan tai
vaikeaan krooniseen invaliditeettiin ja ne kohtaavat lähinnä lapsia
ja nuoria. 1980-luvulta lähtien on tiettyjä lysosomaalisia tauteja
pystytty hoitamaan hematogeenilla kantasolutransplantaatiolla, jolla
saadaan korvattua puuttuva entsyymi. 1990-luvulta lähtien on voitu
valmistaa entsyymiä ja antaa sitä lääkkeenä. Kun on alettu osata
valmistaa poistogeenisiä hiiriä, tämä tekniikka on johtanut
moniin hyviin mahdollisuuksiin testata erilaisia hoitomalleja.
Varhainen diagnostiikka , vastasyntyneitten seulontatutkimukset ja
uudet terapiat ovat kehittelyssä.
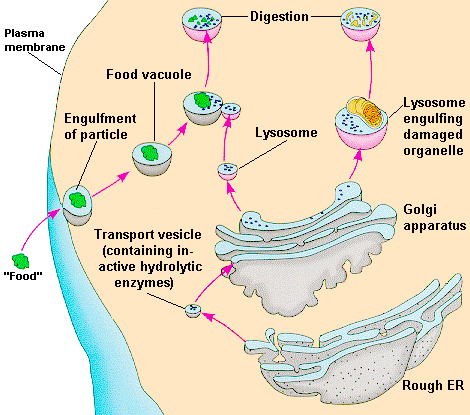
ovat
pieniä yksikköjä tai organelleja, joita esiintyy kehon kaikissa
soluissa paitsi erytrosyyteissä. Niitä kuvasi Christian de Duve
ensi kerran 1950-luvun lopulla . Hän sai vuonna 1974 Nobelpalkinnon
fysiologisesta lääketieteestä tästä löydöstään. Hän kuvasi
myös lysosomien funktiota. Niiden päätehtävänä n hajoittaa
kehon makromolekyylejä matalamolekulaarisiksi yhdisteiksi, joita
metaboloidaan tai hyödynnetään solujen uutissynteesiin
Lysosomeissa
on noin 50 erilaista entsyymiä, joilla on happamassa miljöössä
kykyä hydrolysoida molekulaarisia yhdisteitä, kuten lipidejä,
glykolipidejä, glukosaminoglykaaneja, glukoproteiineja ym.
Geenimutaatiot niissä geeneissä, jotka koodaavat näitä entsyymejä
tai lysosomien kuljetusproteiineja, aiheuttavat synnynnäisiä
aineenvaihdunnallisia tauteja tai synnynnäisiä
aineenvaihdunnallisia virheellisyyksiä.
{kind=link}
{kind=link}
Useita
tämän ryhmän tauteja kuvattiin jo 1800-luvun lopulla ja 19+00
luvun alkupuoliskolla.
Niille annettiin useimmiten nimi sen lääkärin mukaan, jka ensiksi kuvasi kliinisen taudin. Käsitteen lysosomaaliset taudit kiteytti 1960-luvulla Duvesin oppilas niitten sairauksien osalta, joiden syy voitiin johtaa lysosomaalisen entsyymin toiminnan vajauteen. Nykyisin on kuvattuna noin 50 erilaista lysosomaalista tautia, jotka periytyvät pääasiassa autosomaalisesti ja resessiivisti. Muutama tauti periytyy X-kromosomivälitteisesti. Taudit ovat sinänsä harvinaisia, muta ylsiesti ottaen sairausryhmänä koskevat noin 1 lasta 5000 vastasyntneestä. Osa sairaudesta on tavallisempia tietyillä geograafisilla alueilla Ruotsissa tai tietyissä väestöryhmissä. Usea näistä taudeista alkaa elämän varhaisvaiheissa ja johtaa kuolemaan tai vaikeaan invaliditeettiin jo lapsuusiässä. Lapset ovat aluksi terveitä, mutta saavat oireita yhdestä tai useammasta elimestä eri iässä ja eri nopeudella riippuen siitä mikä sairaus niilläon. jokaisessa yksittäisessä taudissa on suuria variaatioita, infantiilit, juveniilit ja aikuismuotonsa riippuen siitä iästä, missä tauti puhkeaa. Joka taudissa on kyse suuresta mutaatiomäärästä. Genotyypin ja fenotyypin välisiä korrelaatioita nähdään harvoin lysosomaalisissa taudeissa, mikä merkitsee vaikeuksia antaa varmaa prognoosia yksittäisissä tapauksissa geenianalyysiin perustamalla. Kaikissa tautitapauksissa voidaan suorittaa seuraavalle sisarukselle prenataalidiagnostiikkaa. Se tehdään joko entsyymidiagnostiikalla tai DNA-analyysillä. Perheen täytyy sitä ennen selvittää periytyvyydet hyvin. selvitelty
Niille annettiin useimmiten nimi sen lääkärin mukaan, jka ensiksi kuvasi kliinisen taudin. Käsitteen lysosomaaliset taudit kiteytti 1960-luvulla Duvesin oppilas niitten sairauksien osalta, joiden syy voitiin johtaa lysosomaalisen entsyymin toiminnan vajauteen. Nykyisin on kuvattuna noin 50 erilaista lysosomaalista tautia, jotka periytyvät pääasiassa autosomaalisesti ja resessiivisti. Muutama tauti periytyy X-kromosomivälitteisesti. Taudit ovat sinänsä harvinaisia, muta ylsiesti ottaen sairausryhmänä koskevat noin 1 lasta 5000 vastasyntneestä. Osa sairaudesta on tavallisempia tietyillä geograafisilla alueilla Ruotsissa tai tietyissä väestöryhmissä. Usea näistä taudeista alkaa elämän varhaisvaiheissa ja johtaa kuolemaan tai vaikeaan invaliditeettiin jo lapsuusiässä. Lapset ovat aluksi terveitä, mutta saavat oireita yhdestä tai useammasta elimestä eri iässä ja eri nopeudella riippuen siitä mikä sairaus niilläon. jokaisessa yksittäisessä taudissa on suuria variaatioita, infantiilit, juveniilit ja aikuismuotonsa riippuen siitä iästä, missä tauti puhkeaa. Joka taudissa on kyse suuresta mutaatiomäärästä. Genotyypin ja fenotyypin välisiä korrelaatioita nähdään harvoin lysosomaalisissa taudeissa, mikä merkitsee vaikeuksia antaa varmaa prognoosia yksittäisissä tapauksissa geenianalyysiin perustamalla. Kaikissa tautitapauksissa voidaan suorittaa seuraavalle sisarukselle prenataalidiagnostiikkaa. Se tehdään joko entsyymidiagnostiikalla tai DNA-analyysillä. Perheen täytyy sitä ennen selvittää periytyvyydet hyvin. selvitelty
Lysosomaaliset
taudit jaetaan alaryhmiin, jotka viittaavat siihen substanssiin,
mitä keho ei pysty hajoittamaan (ESIM: Mukopolysakkaroosi I) .
Alaryhmät sisältävät vuorostaan useita yksittäisiä tauteja.
Sen lisäksi, että taudilla on keksijänsä nimi (tässä
esimerkissä Hurlerin tauti), niillä on nykyisin myös nimi
entsyymipuutteen mukaan, tässä esimerkissä alfa-1-L-iduronidaasin
puute. Useat näistä lysosomaalisista taudeista löytyy
ruotsinkielisessä lähteessä
Socialstyrelsens webbplats, http://www.sos.se/smkh .
Socialstyrelsens webbplats, http://www.sos.se/smkh .
Luonteenomaista
useimmissa mukopolysakkaridooseissa (MPS) glukosaminoglykaanien(GAG)
kertyminen aiheuttaa karkeita kasvonpiirteitä, lyhytkasvuisuutta ja
luustovaikutusta, niveljäykistymiä, hepatosplenomegaliaa ja
keskushermoston vikuuntumista ja samalla mentaaliset kyvyt
heikkenevät, näkökyky, kuulo myös vaikuttuvat eri asteisesti
riippuen taudin tyypistä.
MPS
I, Hurlerin taudissa lapset tuskin saavuttavat 10 vuoden ikää.
Vuonna 2007 oli noin 40 henkilöllä MPS-tauti diagnosoituna.
noin yksi lapsi vuodessa syntyy Hurlerin taudissa.
Lipidooseissa,
missä sfingomyeliini, gangliosidi tai cerebrosidi ei hajoa
täydellisesti, kärsii lähinnä hermojärjestelmä. Tärkeimmät
oireet ovat mentaalisten ja motoristen valmiuksien katoaminen,
pareesien kehittyminen, mentaaliretardaatio, näön kadottaminen sekä
epilepsia.
Krabben
taudissa lapsi tulee enemmän ja enemmän spastiseksi 3-5 kk iässä
ja kuolee parissa vuodessa. Vuosittain syntyy 4 lasta Krabben
tautisena.
Tässä
taudissa lapset kehittyvät normaalisti noin 14-15 kk ikään ja
siten kadottavat nopeasti valmiuksiaan, savat vaiketia kipuja ja
harvoin elävät yli 10 vuotiaiksi. Rtosissa syntyy n. 2 lasta
vuodessa tässa taudissa.
Tavallisinta
Ruotsissa on lapsuusiän varhain alkava ja nopeasti etenevä Krabben
tauti, metakromaatinen leukodystrofia (MLD) ja Gaucherin tauti tyyppi
III, Norrbotten-muoto. Gaucherin taudissa III-tyypissä on
hepatosplenomegalia usein ensimmäinen diagnostisoitu poikkeus, kun
taas hematologisia ja kerebraalisia oireita tulee myöhemmin. Noin 50
henkilöä Ruotsissa potee erilaisia Gaucherin taudin lajeja.
aiheuttaa
tunnistettavissa olevia oireita ja sitä identifioidaan vasta
aikuis-iässä. Tässä taudissa vikuuntuu lähinnä verisuonet,
munuaiset ja sydän ja siitä seuraa varhainen halvaus ja
munuaisinsuffisienssi. noin 40 henkilöä on diagnostisoitu
Ruotsissa.
Tässä
glykoproteinoosissa on tyypilliset kasvonpiirteet ,
lyhytkasvuisuutta, mentaalista retardaatiota ja nivelten jäykkyyttä
ja aspartyyliglukosaminuriaa. Tätä on suomalaisperäisessä
väestössä Ruotsissa.
Toinen
tauti, jossa on defekti glukosidaasientsyymi, on Pompen tauti,
glykogenoosi tyyppi II , jonka kuvasi Hers 1961. Siinä tapahtuu
glykogeenin latautuminen lihaksistoon. Sydänlihas voi vikuuntua niin
pahasti infantiilissa muodossa, että lapsi kuolee pikkulapsi-iässä,
kun taas myöhemmässä iässä lähinnä ilmenee progredioivaa
lihasheikkoutta. Harvalla diagnosoidan pompen tautioa Ruotsissa.
Kuljetusdefekteistä
on tavallisin tauti Ruotsissa ns sallan tauti. Se progredioituu
hitaasti ja siihen kuuluu ataksia, mentaali retardaatio ja lähes
normaali elinikä. noin 30 henkilöä omaa tämän diagnoosin
Ruotsissa tällä hetkellä.
Tämä
tauti luetaan myös lysosomaalisiin tauteihin ja jaetaan useisiin
alaryhmiin siirppuen siitä, missä iässä tauti alkaa ja mikä on
elinikä.
Tämä on
Ruotsissa tavallisinta ja joka vuosi diagnosoidaan 2-3 lasta. Tämä
tauti voi kohdata varhaisessa kouluiässä ja sen etenemiseen kuuluu
näkökyvyn katoaminen, henkisten kykyjen huononeminen ja puheen
huononeminen, lisääntyviä halvauksia ja epilepsiaa.
Muut ovat
varsin harvinaisia Ruotsissa.
Kliininen
kuva
Virtsanäyte
Valkosolujen
entsyymianalyysi
Fibroblastianalyysi
Mutaatiodiagnostiikka
Kantajuusdiagnostiikka
Prenataalidiagnostiikka
Neonataali
seulonta (Krabben tauti)
Entsyymiaktiviteetit,
entsyymiproteiinit, kertymätuotteet
PKU testi
Siirretaan kantasoluja : hematogeeni kantasolutransplantaatio ( varsinkin lysosomaalisiin ja peroksisomaalisiin tauteihin: siirtosolut tuottavat puuttuvaa entsyymiä luuyrtimessä). Entsyymi siirtyy eri elimiin veren tai makrofagien ( Kuppferin solujen, mikrogliasolujen) avulla. Mikrogliat pääsevät aivojen puolelle ja voivat tuottaa entsyymiä aivoissa, joskin vain pieniä määriä. Pienillä lapsilla voi käyttää napanuoraverta kantasolulähteenä perinnöllisen ainenvaihduntataudin hoidossa. Siinä on enemmän kantasoluja kuin luuytimessä. Efekti luustoon ja myeliiniin voi olla tehokkaampi niistä käsin.Napanuoraperäisten verisolujen annossa on haittana pieni annos. Jos donorina on HLA-identtinen sisarus, mortaliteetti on matalin. Hurlerin taudissa tai MPS I tyypissa on paras siritohetki 18-24 kk ikä. krabben taudissa napanuoraverisoluje siirto suoritetaan ennen 40 päivän ikää. Siirtoja suositellaan myös MPS VI, MLD juveiniilissa ja aikuismuodossa, Krabben taudissa mannosidoosissa, Niemann–Picks A ja B taudissa sekä AGU sairaudessa.
Mesenkymaalisia
(pluripotentteja) kantasolujakin on siirretty (Hurler, MLD).
Vektorina
adenovirus tai retrovirus on saatu eläinmallissa hyviä tuloksia.
Gaucherin
T III taudissa 1990 luvun alusta. Aiemmin oloi transplantaatio ainoa
mahdollisuus.
Entsyymiterapiaa
on annettu seuraavissa. Pompen tauti, Fabryn tauti , myöhään
alkava MPS I, MPS II ja MPS VI.
On menossa
tutkimuksia MLD, MPS IV ja MPS III A suhteen.
Terapia
annetaan suoneen joka viikko tai joka toinen viikko. Entsyymit eivät
pääse aivon puolelle. Kallista terapiaa. Joissain taudeissa
annetaan intratekaalista entsyymiä.
Patologisesti
kertyvän aineksen biosynteesiä vähennetään.Tällainen kokeilu on
menossa Gaucherin taudin suhteen.
Sitä
entsyymiä, jota on, aktivoidaan lisäämällä pieniä molekyylejä
ns. chaperoneja. Poistogeenihiirillä testataan
erilaisia hoitomalleja. N-butyylideoxinorijimyciini vähentää
kertyvän substaatin synteesiä. Se toimii kuin chaperoni. Defekti
entsyymi ei hajoa niin nopeasti ja sitä voidaan ottaa lysosomiin ja
siten se voiehtiä olla aktiivi. Entsyymiä voidaan myös
stabilisoida.
{kind=link}
Inga kommentarer:
Skicka en kommentar